order histories, retained contact details for faster checkout, review submissions, and special promotions.
Forgot password?
order histories, retained contact details for faster checkout, review submissions, and special promotions.
Locations
Orders Processing,
Shipping & Receiving,
Warehouse
2 Shaker Rd Suites
B001/B101
Shirley, MA 01464
Production Lab
Floor 6, Suite 620
20700 44th Avenue W
Lynnwood, WA 98036
Telephone Numbers
Tel: +1 (206) 374-1102
Fax: +1 (206) 577-4565
Contact Us
Additional Contact Details
order histories, retained contact details for faster checkout, review submissions, and special promotions.
Forgot password?
order histories, retained contact details for faster checkout, review submissions, and special promotions.
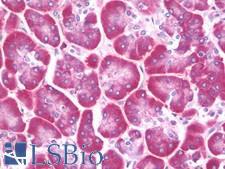
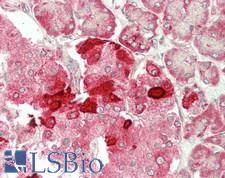
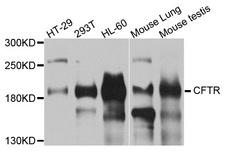
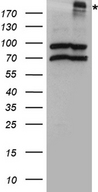
CFTR
cystic fibrosis transmembrane conductance regulator (ATP-binding cassette sub-family C, member 7)
CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), sometimes also referred as ABCC7 (ATP-binding cassette, Subfamily C, Member 7), is a member of the ATP binding cassette (ABC) transporter family. CFTR functions as a chloride channel and controls the regulation of other transport pathways. Homozygous mutations in the CFTR gene cause cystic fibrosis (CF), formerly known as mucoviscidosis, a common hereditary disorder characterized by severe abnormalities in respiratory, digestive and other organ systems. Approximately 70% of the mutations in CF patients correspond to a specific deletion of 3 base pairs, which results in the loss of a phenylalanine at position 508. The approaches aimed to restore the CFTR gene function in CF patients are the subject of intensive research and include the development of gene therapy protocols. In addition to CF syndrome, CFTR mutations were found in patients suffering from bilateral aplasia of the vas deferens (CBAVD). The high frequency of CFTR defects in the human population can be explained by increased resistance to infectious diseases in heterozygous mutation carriers. Alternative splice variants have been described, many of which result from mutations in the CFTR gene.
| Gene Name: | cystic fibrosis transmembrane conductance regulator (ATP-binding cassette sub-family C, member 7) |
| Family/Subfamily: | Transporter , ATP-binding cassette - ABCC/MRP |
| Synonyms: | CFTR, ABC35, ABCC7, CF, DJ760C5.1, MRP7, CFTR/MRP, TNR-CFTR |
| Target Sequences: | NM_000492 NP_000483.3 P13569 |
Publications (2)
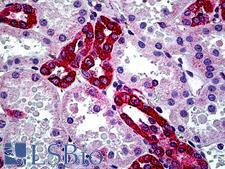

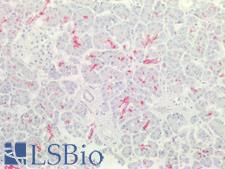
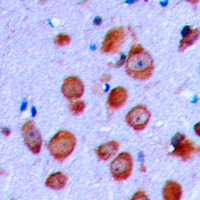
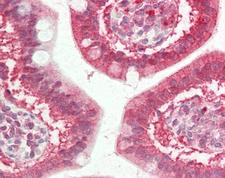





If you do not find the reagent or information you require, please contact Customer.Support@LSBio.com to inquire about additional products in development.



